Research in our lab
Our lab is broadly interested in the evolutionary processes linked to adaptation of species and the composition of genomes. We attempt to span a wide range of scales, from population-level investigations to analyses covering large taxonomic groups. A primary focus is the threat of pathogens in agriculture. Genetic homogeneity of crop plants and antifungals drive rapid adaptive evolution in pathogen populations. We use a combination of genomics and experimental approaches to dissect adaptation, from the emergence of new pathogens to the evolution of fungicide resistance. We are also interested in the evolutionary processes that shape the composition of genomes, including the role of transposable elements and other selfish DNA sequences in genome evolution. Finally, we are passionate about applying conservation genomics to understand the evolutionary trajectory of endangered species.

Our main model is a worldwide pathogen of wheat called Zymoseptoria tritici, which causes the most economically damaging foliar disease in Europe. The pathogen has overcome most resistance of the host and all currently available fungicides. Genomes of this pathogen show an extraordinary level of sequence and chromosomal polymorphism unlike many other pathogens. One of our major aims is to understand how major adaptive traits evolved. For this, we perform genome-wide association studies (GWAS) to map phenotypic traits to individual loci in the genome. We focus in particular on different components of pathogenicity but are also interested in stress tolerance against fungicides, temperature and conditions experienced during infection (low pH, oxidative stress). To understand the process of adaptive evolution, we analyse major effect loci for signatures of selection and sequence rearrangements.

We are fascinated by the rapid dynamics of transposable elements (TE) in genomes. We believe that the compact yet dynamic genomes of fungal plant pathogens are quite unique models to address fundamental questions about the genome biology of TEs. We focus on what facilitates and constrains TE evolution in genomes by analyzing the shortest possible time scales (i.e. within species, populations or pedigrees). We investigate some highly active TEs that simultaneously made contributions to recent adaptation, the instability of chromosomes and the expansion of genomes. We also use large-scale screens of trait variation among individuals to associate TE dynamics to recent adaptation.
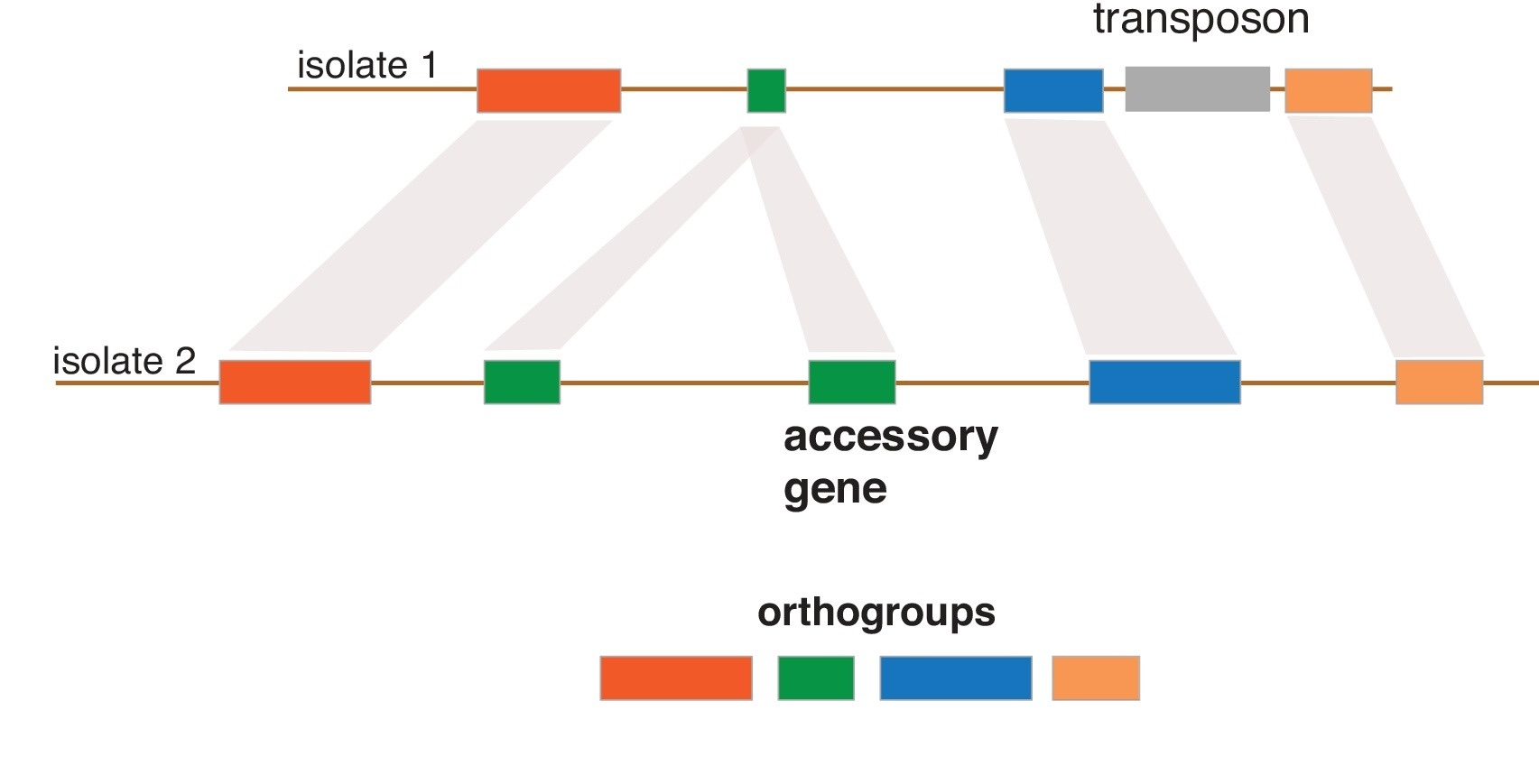
Our group is also interested in understanding the evolution of genome architecture. In particular, we want to retrace the emergence of novelty (e.g. new genes) and major structural rearrangements. For this, we assemble and analyse complete fungal genomes. We found that homologous Z. tritici chromosomes segregate significant structural rearrangements within populations and that such variation created key adaptations to overcome host resistance. The genome of Z. tritici also comprises a highly polymorphic complement of eight chromosomes that are not found in every member of the species. During meiosis, accessory chromosomes frequently undergo rearrangements, fusions and non-disjunction events. Our aims are to precisely identify the mechanisms responsible for the chromosomal abberrations and how chromosomal polymorphism is generated and maintained in populations.
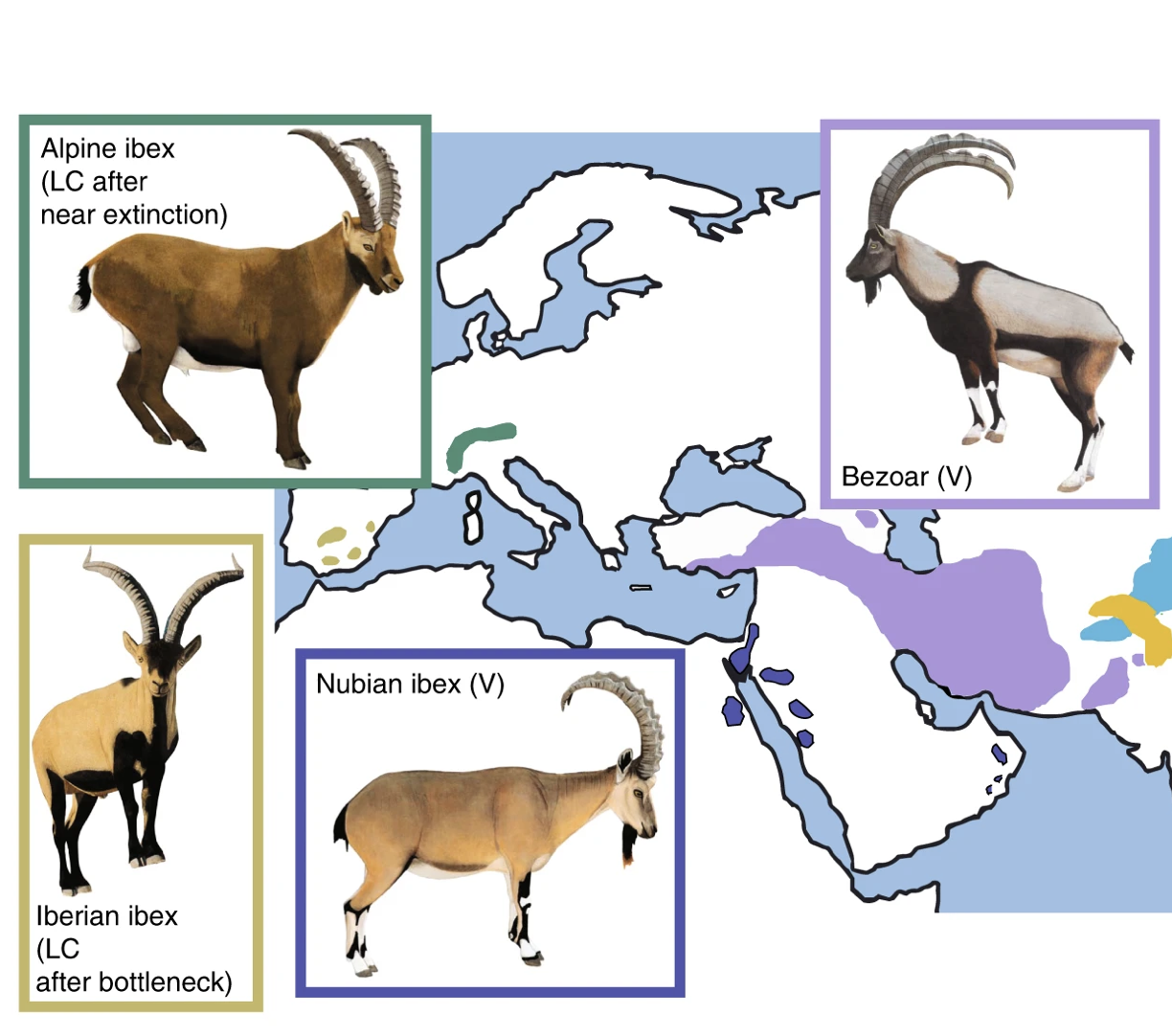
Our group collaborates on research projects on Alpine ibex and other flagship species threatened by small population size. Alpine ibex suffered from a population bottleneck of a few hundred individuals caused by overhunting. The species was reestablished across the Alps and counts again around 40’000 individuals. We showed that Alpine ibex received genetic material from domestic goats through hybridization and introgression. A process that was likely favored by the population bottleneck. We are also interested in the consequences of population bottlenecks on the fate of deleterious mutations.